Lab 1 Report
Class Diagram
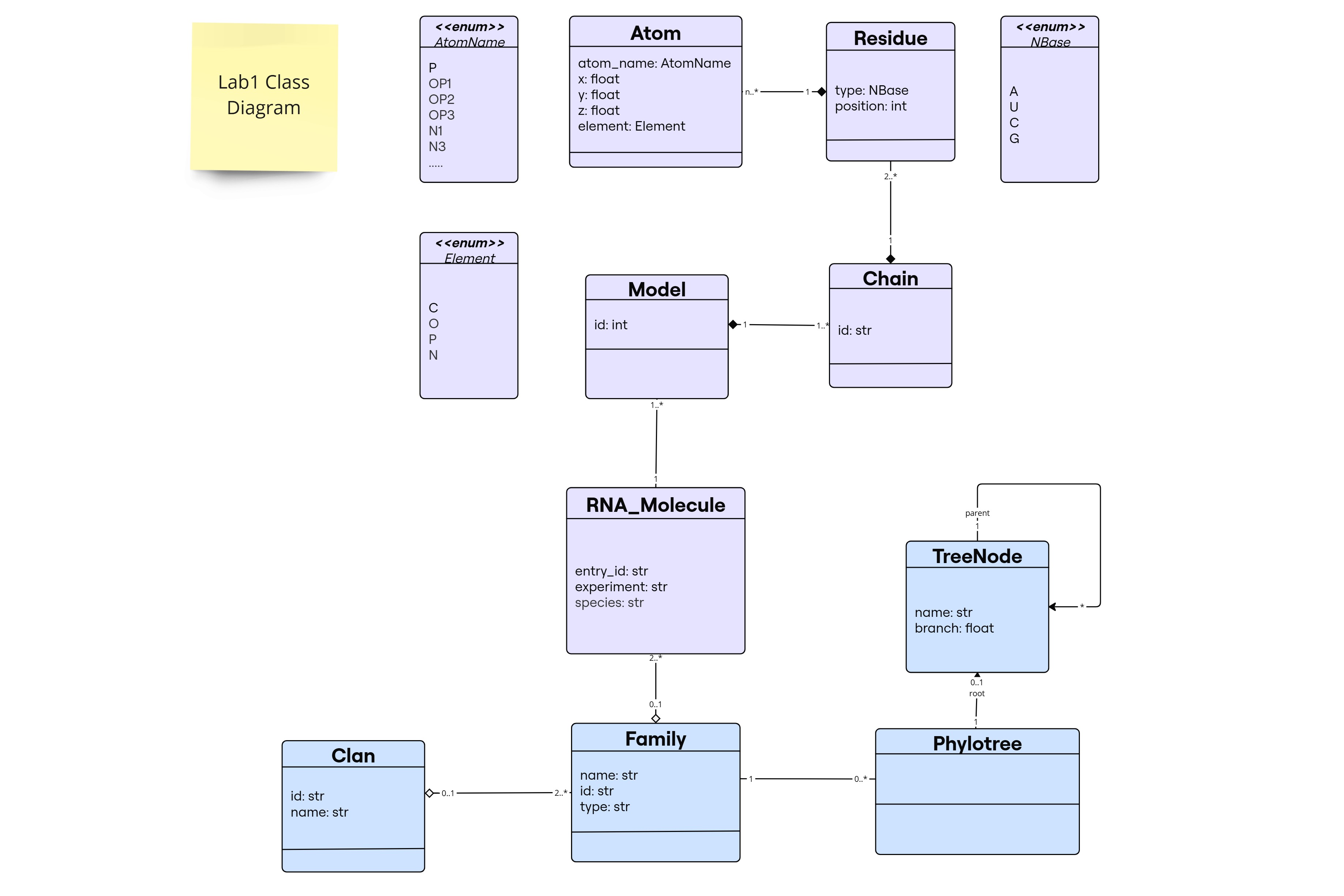
According to this lab description, the first part is about RNA sequences and their spatial conformations. From the details and examples provided, we assumed that the RNA sequence that we want to model has a structure and the purpose is the manipulation of this structure.
Entity Atom
- Attributes: atom_name: AtomName, x: float, y: float, z: float, element: Element
- AtomName is an enumaration that contains all the possible atom names as present in pdb.
- Element is an enumeration that contains all the possible atoms that can be present in an RNA nucleotide “C, O, P, N”.
- x y z are the coordinates of the atom in the 3D space.
Entity Residue
- Attributes: type: NBase, position: int
- NBase is an enumaration that contains all the possible residue types for RNA nucleotides “A, U, G, C”.
- position is the position of the residue in the RNA sequence.
- There is a composition relationship between Residue and Atom. 1 Residue is composed of multiple atoms. If a Residue is deleted, all the atoms that are part of that residue will be deleted as well.
Entity Chain
- Attributes: id: str
- id is the identifier of the chain.
- There is a composition relationship between Chain and Residue. 1 Chain is composed of multiple residues (2 residues at least since a residue is a nucleotide that is linked by a phosphodiester bond to another nucleotide, even though of course it will be formed of many more residues). If a Chain is deleted, all the residues that are part of that chain will be deleted as well.
Entity Model
- Attributes: id: int
- id is the identifier of the model.
- We considered a model as a specific conformation of an RNA molecule (or structure).
- There is a composition relationship between Model and Chain. 1 Model is composed of at least one chain. If a Model is deleted, all the chains that are part of that model will be deleted as well.
Entity RNA_Molecule
- Attributes: entry_id: int, experiment: str, species: str
- Since we considered that we are modeling only RNA molecules that have experimental structures, we defined attributes entry_id as in pdb entry id, experiment as the experiment that was performed to obtain the structure and species as the species source of where this RNA molecule was obtained from.
- An RNA_Molecule can have 1 or multiple models. If it was determined by X-Ray it will generally have only one model, but if it was determined by NMR it will have multiple models. And so for example if this RNA_Molecule was determined by NMR, we can access all the models that were generated from this NMR experiment, each model indexed by a number.
Entity Family
- Attributes: name: str, id: str, type: str
- There exist an aggregation relationship between Family and RNA_Molecule. 1 Family consists of evolutionary related RNA sequences sharing common similarities. So a family should at least contain 2 RNA molecules that are similar. But there might exist an RNA molecule that is not part of any family maybe because it is unique or because it is not yet classified. It is aggregation relationship because we can assume that if a family is deleted the RNA molecules will still exist.
Entity Clan
- Attributes: name: str, id: str
- Same logic as Family, but a Clan is a group of families that share common characteristics. So it will be an aggregation relationship and 1 clan consists of at least 2 families. But a family can exist without being part of any clan.
Entity PhyloTree
- Since a phylogenetic tree here is defined for a given RNA family, hence a tree belongs only to one family.
- But a family can have 0 (if it does not yet have a tree associated with it) or multiple phylogenetic trees (because probably different algorithms can be used to construct the tree, or different versions at different times can exist).
- An important consideration during the uml design is portraying teh class as a Tree data structure which by definition is going to be recursive, this is embedded through the
rootrelationship in thePhyloTreeclass withTreeNode, that will be represented as an attribute during implementation.
Entity TreeNode
[!NOTE] This class is a helper class for PhyloTree. While it’s not explicitly mentioned in the description, we decided to include it in the model to represent the nodes in the tree. It was a crucial addition in order to describe the class a tree data structure and allow for the implementation of the tree traversal methods.
The TreeNode class serves to represent nodes in a phylogenetic tree within Pylotree, hence Phylotree has attribute of type TreeNode. It functions as the fundamental unit of the nodes list attribute, storing information in a graph-based data structure (by having a recurive link to other nodes, parent and children, in its attributes). Each node holds an RNA type as its data attribute and maintains a list of child nodes, this is shown through the self relationship in the class diagram, where a node can have multiple children nodes. Key attribute is branch_length, which represents the distance to the parent node.
Species
It is described that species refer to the organisms that contain RNA sequences belonging to a particular RNA family. So a family can be distributed across multiple species, and a species can contain multiple families. But we did not include it as a separate class but rather as an attribute of RNA_Molecule, and so we can obtain the distribution of the species for a particular family by looking at the species attribute of the RNA_Molecules that are part of that family. And in this way, the species description can also be used in the phylogenetic tree.
Object Diagram
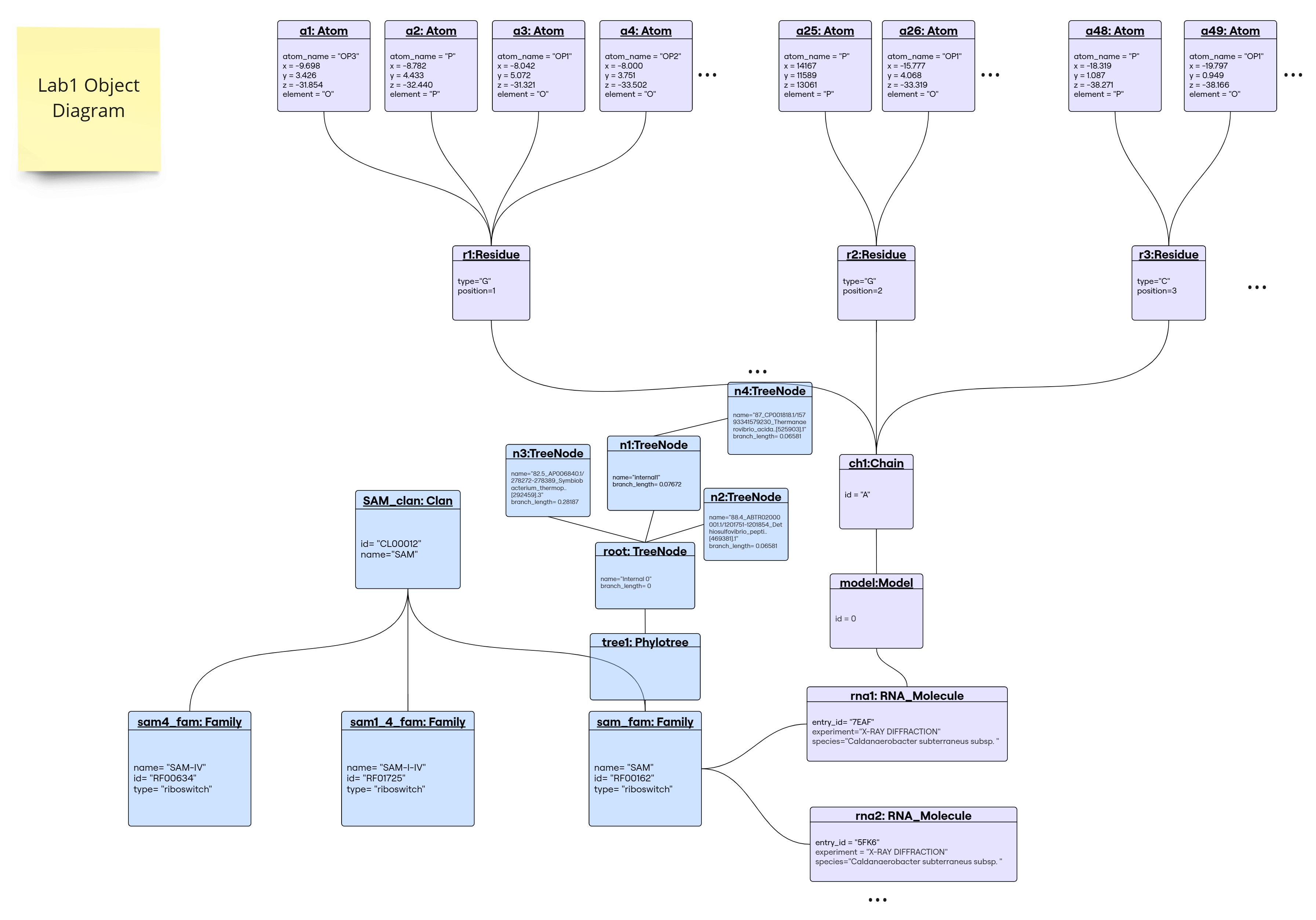
This object diagram portrays the example found in here. It shows the following objects:
| object | class | description |
|---|---|---|
| a1 | Atom | An oxygen atom labeled “OP3” with specific coordinates (3rd oxygen of a phosphate molecule from a residue r1, found through the link) |
| a2 | Atom | A phosphorus atom labeled “P” with specific coordinates. |
| a3 | Atom | An oxygen atom labeled “OP1” with specific coordinates. |
| a4 | Atom | An oxygen atom labeled “OP2” with specific coordinates. |
| a25 | Atom | A phosphorus atom labeled “P” positioned further in the structure. |
| a26 | Atom | An oxygen atom labeled “OP1” linked to atom a25. |
| a48 | Atom | A phosphorus atom labeled “P” in another part of the structure. |
| a49 | Atom | An oxygen atom labeled “OP1” linked to atom a48. |
| r1 | Residue | A guanine residue positioned first in the sequence (has linked atoms a1, a2, a3…) |
| r2 | Residue | A guanine residue positioned second in the sequence. |
| r3 | Residue | A cytosine residue positioned third in the sequence. |
| ch1 | Chain | A chain labeled “X” that links residues together. |
| model | Model | The structural model representation, labeled with ID 0 (since x-ray structure, normally structure is one model, 0) |
| rna1 | RNA_Molecule | An RNA molecule identified by entry “7EAF,” studied via X-ray diffraction. |
| rna2 | RNA_Molecule | Another RNA molecule identified by entry “5KF6,” analyzed similarly. |
| sam_fam | Family | a SAM riboswitch family named “SAM” which is the one 7eaf molecule belong to, thus the relationship |
| sam1_4_fam | Family | SAM riboswitch family named “SAM-I/IV”, belonging to SAM clan |
| sam4_fam | Family | a riboswitch family named “SAM-IV”, belonging to SAM clan |
| sam_clan | Clan | clan of riboswitch families named “SAM”, linked to 3 families that belong to it |
| tree1 | Phylotree | phylogenetic tree representation of related sequences for the family SAM (thus the link) |
| root | TreeNode | The root node of the phylogenetic tree, attribute of tree1, thus the link with the PhyloTree object, it’s connected to all other through parent-child links |
| n1 | TreeNode | An internal tree node, conceptually a common ancestor, with a specific branch length away from root (since its linked directly to root) |
| n2 | TreeNode | tree node representing a RNA molecule of a particular species (leaf) |
| n3 | TreeNode | Another tree node representing a RNA molecule of a particular species |
| n4 | TreeNode | Another leaf representing a rna seq labeled with accession “92. CP018551.1” |
Python Implementation
List of modules:
Atom.pyResidue.pyChain.pyModel.pyRNA_Molecule.pyFamily.pyClan.pyPhyloTree.pyutils.pythis includes helper functions for database calls, data extraction, and file handling…
Class Atom
- It includes the attributes defined in the class diagram, with enum implementation for
AtomNameandElementfor validation. - In the constructor, the
atom_nameandelementattributes are string values, but they were later converted to the corresponding enumeration values in the setter methods. @attribute.setteris used to validate the input values for the attributes. For the coordinates it validates that the values are float, and for the atom_name and element it validates that the values are part of the enumeration. For example:def element(self, element): if not isinstance(element, str): raise TypeError(f"element must be a string, got {type(element)}") #Check if the string is a valid Element if not element in Element.__members__: raise ValueError(f"{element} is not a valid Element value") self._element=Element.__members__[element]It is used to validate that the input value is a string and that the string is part of the Element enumeration.
@propertydecorator was used to define getter and setter methods for the attributes, allowing for encapsulation and validation while providing a simple interface for accessing and modifying the attributes.__repr__is used to return the string representation of the object as:atom_name x y z element.
Class Residue
- It includes the attributes
typeandposition, with enum implementation forNBasefor validation of the residue type. - In the constructor, the
typeattribute is a string value, but it was later converted to the corresponding enumeration value in the setter method. It also includes ` atoms=None` as a default value for the atoms attribute, which is a list that will store the atoms that are part of the residue, it is initialized as an empty list if no atoms are provided. @propertyand@attribute.setterare used for the attributes, with setter used for validation of the input values.- Methods for adding
add_atom()with validations that the input is an instance ofAtomand that it does not already exist and removing atomsremove_atomfrom the residue are included, with a method to also get the list of atoms in the residueget_atoms(). __repr__is used to return the string representation of the object as:type position.
Class Chain
- It includes the attribute
id, withresidues=Noneas a default value for the residues attribute, which is a list that will store the residues that are part of the chain, it is initialized as an empty list if no residues are provided. - Methods for adding
add_residue()with validations and removing residuesremove_residuefrom the chain are also included, with a method to get the list of residues in the chainget_residues(). __repr__is used to return the string representation of the object as:id.
Class Model
- It includes the attribute
id, withchains=Noneas a default value for the chains attribute, which is a list that will store the chains that are part of the model, it is initialized as an empty list if no chains are provided. - Methods for adding
add_chain()with validations and removing chainsremove_chainfrom the model are also included, with a method to get the list of chains in the modelget_chains(). __repr__is used to return the string representation of the object as:id.
Class RNA_Molecule
- It includes the attributes
entry_id,experiment,species, withmodels=Noneas a default value for the models attribute, which is a list that will store the models that are part of the RNA_Molecule, it is initialized as an empty list if no models are provided. - Methods for adding
add_model()with validations and removing modelsremove_modelfrom the RNA_Molecule are also included, with a method to get the list of models in the RNA_Moleculeget_models(). __repr__is used to return the string representation of the object as:entry_id experiment species.@propertyand@attribute.setterare used for the attributes, with setter used for validation of the input values.print_all()method is used to print all the models, chains, residues, and atoms that are part of the RNA_Molecule similar to a pdb file format, and saves the output to a file. The format is as follows:ATOM <atom_number> <atom_name> <residue_type> <chain_id> <residue_position> <x> <y> <z> <element>
Class Family
FamilyClass Overview:
The Family class represents a family of RNA molecules, particularly those in the Rfam database. It ensures that each family is uniquely identified, maintains a list of RNA molecules as its members, and optionally includes a phylogenetic tree representation. The class prevents duplicate instances and provides structured methods for adding, removing, and retrieving RNA families.
Key Features
- Represents RNA families with shared sequence similarity, secondary structure, and function.
- Ensures uniqueness by preventing duplicate instances based on the family ID
- Supports integration with the Rfam database for automatic family creation (in
utils.py, using rfam api) - Includes a phylogenetic tree (
Phylotree) to represent evolutionary relationships which can be retrieved from various data types: newick, dict and json.
Attributes
Class Attributes
entries: List of all createdFamilyobjects to track and avoid duplicates.
Instance Attributes
id(str): Unique identifier for the family.name(str): Name of the RNA family.type(str, optional): Type of RNA (e.g., rRNA, tRNA, miRNA).members(list): List ofRNA_Moleculeobjects representing the
helper methods (private):
- __validate_member(member): validate if the member is an instance of RNA_Molecule
- __delete_family(id): delete the family with the given id from the entries list
dunders:
__init__(id, name, type=None, members=[], from_database=False): from_database is a flag to indicate if the object is created using the generator function from the database__del__(self)__eq__(self, other)__len__(self)__getitem__(self, key)__setattr__(self, name, value)__str__(self)__repr__(self)
Class Clan
Clan Class Overview
The Clan class represents a group of RNA families that share common ancestry or biological significance. It ensures unique identification of clans, prevents duplicates, and provides structured methods for managing RNA families (Family objects).
Key Features
- Represents RNA family groups with shared ancestry.
- Prevents duplicate clan creation by linking existing clans if an ID already exists.
- Provides methods to add and remove RNA families (
Familyobjects). - Implements custom string representation for better readability.
Class Attributes
entries: List of all createdClanobjects to track and avoid duplicates.
Instance Attributes
id(str): Unique identifier for the clan (immutable).name(str, optional): Name of the clan.members(list): List ofFamilyobjects that belong to the clan.
Class Methods
get_instances(): Returns a list of all createdClanobjects.get_clan(id): Retrieves an existing clan by its ID.
Instance Methods
add_family(family): Adds aFamilyobject to the clan.remove_family(family): Removes aFamilyobject from the clan.
Private Methods
__validate_member(member): Ensures that onlyFamilyobjects are added to the clan.
Magic Methods
__str__(): Returns a formatted string representation of the clan and its families.__repr__(): Returns a structured representation of the clan instance.__eq__(other): Checks equality based on clan ID.__setattr__(name, value): Ensures controlled attribute setting, preventing ID modification and enforcing type validation.
Class PhyloTree and TreeNode
This module tree.py defines a TreeNode class and a Phylotree class for constructing and managing a phylogenetic tree.
In the object diagram we see a phylotree is diretly linked to one node, which is the root (will be portrayed as an attribute) and each node is recursively linked to other nodes, in its attributes, as seen in the following implementation:
The TreeNode class represents a node in the tree with attributes:
name: stores the RNA type.branch_length: stores the distance to the parent node.parent: stores the parent node.children: stores child nodes as a dictionary.
Methods in TreeNode:
add_child(child, weight): adds a child node with a given branch length.preorder_traversal(level=0): performs a preorder traversal and returns a string representation.__repr__and__str__: provide readable representations of the node.__getitem__: retrieves a child node by name.
The Phylotree class represents a phylogenetic tree for RNA sequences, constructed using computational phylogenetics. It consists of:
- A
rootnode. - Internal nodes representing common ancestors.
- Leaf nodes representing specific RNA sequences.
Methods in Phylotree:
build_tree(tree_dict, parent=None): builds a tree from a dictionary.from_dict(tree_dict, parent=None): constructs a tree from a dictionary.from_json(json_str): constructs a tree from a JSON string or file.from_newick(newick_str): parses a Newick-formatted string or file to build the tree.__str__and__repr__: return a string representation of the tree.
The module demonstrates tree construction using different input formats:
- A dictionary representation.
- A JSON file.
- A Newick-formatted string (or newick file path)
Example usage:
tree_dict = {
"children": [
{"name": "a", "branch_length": 0.05592},
{"name": "b", "branch_length": 0.08277},
{
"children": [
{"name": "c", "branch_length": 0.11049},
{"name": "d", "branch_length": 0.31409}
],
"branch_length": 0.340
}
],
"branch_length": 0.03601
}
tree = Phylotree.from_dict(tree_dict)
print(tree)
or
newick_str = '''
(87.4_AE017263.1/29965-30028_Mesoplasma_florum_L1[265311].1:0.05592,
_URS000080DE91_2151/1-68_Mesoplasma_florum[2151].1:0.08277,
(90_AE017263.1/668937-668875_Mesoplasma_florum_L1[265311].2:0.11049,
81.3_AE017263.1/31976-32038_Mesoplasma_florum_L1[265311].3:0.31409)
0.340:0.03601);
'''
tree=Phylotree.from_newick(newick_str) #success
or from files:
tree=Phylotree.from_newick('lab1/examples/RF00162.nhx') #newick
tree=Phylotree.from_json('lab1/examples/RF00162.json') #json
Functions to Extract Data from PDB Files to Create RNA_Molecule Object
fetch_pdb_file(pdb_entry_id, save_directory=CACHE_DIR)function written inutils.pyis used to fetch the pdb file from the RCSB PDB database using the pdb entry id. It saves the file in the specified directory. It uses theBiopythonlibrary to fetch the file.create_RNA_Molecule(pdb_entry_id)function written inutils.pyis used to create an RNA_Molecule object from the pdb file accessed through the pdb_entry using the first function. It reads the pdb file, extracts the necessary information first about theexperimentandspeciesto create the specificRNA_moleculeobject, and then creates the corresponding objects (models, chains, residues, atoms) while adding them in the hierarchical order to the RNA_Molecule object. It returns the RNA_Molecule object.
Other utility functions
[!IMPORTANT] The
utils.pyfile contains helper functions for database calls, data extraction, and file handling. It includes interaction with the Rfam database API to retrieve information about RNA families and phylogenetic trees, automate etxraction, and manipulate various file formats likenewicktrees. In this module, if any files have to be downloaded as intermediary steps, they are saved in a CACHE directory defaulted to a hidden directory.rnalib_cache/in teh working dir to avoid repeated downloads.
- The
get_rfam(q:str)function is used to get the information about an RNA family from the Rfam database using the family ID. It returns the information in JSON format. - The
get_family_attributes(q:str)function is used to get the name, identity, and curation type of an RNA family from the Rfam database using the family ID. It returns the information as a tuple. - The
get_pdb_ids_from_fam(fam_id)function is used to get the PDB IDs associated with an RNA family from the Rfam database using the family ID. It returns the PDB IDs as a list (helpful to automate the extraction of all RNA molecules found on Rfam from the PDB database). - The
get_tree_newick_from_fam(fam_id)function is used to get the Newick tree from the Rfam database given the RNA family ID. It returns the Newick tree as a string. - The
parse_newick(newick)function is used to parse a Newick string into a nested dictionary. It is used to parse the Newick tree obtained from the Rfam database, using regex to extract the tree structure.
Python Code Examples
Kindly find an example of the implementation of the classes in this notebook
Small examples inside each class implementation:
- Specific Usage Examples are present at the end of each class implementation in the python files.
- The examples demonstrate the creation of objects, adding and removing elements, and printing the objects.
- For example:
- in the
Atomclass:atom = Atom("C1'", 1.0, 2.0, 3.0, "C") print(atom) #output: C1' 1.0 2.0 3.0 C - in the
Residueclass: ```python r = Residue(“A”, 1) print(r) #output: A 1 atom1 = Atom(“C1’”, 1.0, 2.0, 3.0, “C”) atom2 = Atom(“N9”, 4.0, 5.0, 6.0, “N”) r.add_atom(atom1) r.add_atom(atom2) print(r.get_atoms()) #output: [C1’ 1.0 2.0 3.0 C, N9 4.0 5.0 6.0 N] r.remove_atom(atom1) print(r.get_atoms()) #output: [N9 4.0 5.0 6.0 N]
atom3 = Atom(“C4”, 7.0, 8.0, 9.0, “C”) r2 = Residue(“G”, 2) r2.add_atom(atom3) print(r2.get_atoms()) #output: [C4 7.0 8.0 9.0 C]
- in the `Chain` class: ```python c = Chain("A") print(c) #output: A r = Residue("A", 1) c.add_residue(r) print(c.get_residues()) #output: [A 1] c.remove_residue(r) print(c.get_residues()) #output: []- in the
Modelclass:m = Model(1) print(m) #output: 1 c = Chain("A") m.add_chain(c) print(m.get_chains()) #output: [A] m.remove_chain(c) print(m.get_chains()) #output: [] - in the
RNA_Moleculeclass:rna1 = RNA_Molecule("1A9N", "NMR", "Homo sapiens") print(rna1) #Output 1A9N NMR Homo sapiens m1 = Model(1) m2 = Model(2) m3 = Model(3) rna1.add_model(m1) rna1.add_model(m2) rna1.add_model(m3) print(rna1.get_models()) #Output [Model 1, Model 2, Model 3] rna1.remove_model(m3) print(rna1.get_models()) #Output [Model 1, Model 2] rna1.print_all() #Output 1A9N NMR Homo sapiens #Model 1 #Model 2
- in the
Small Example to Manually create an RNA_Molecule
- Provided in the file
Example.py. - It shows how the code can be used to create an RNA_Molecule manually, while adding all its structures from models, chains, residues, and atoms.
#Creating an RNA molecule rna_molecule = RNA_Molecule("1JAT", "X-RAY DIFFRACTION", "Homo sapiens") #Creating a model model1 = Model(1) #Creating a chain ch1 = Chain('A') #Adding the model to the RNA molecule rna_molecule.add_model(model1) #Adding the chain to the model model1.add_chain(ch1) #Creating Residues res1=Residue("A", 1) res2=Residue("U", 2) res3=Residue("C",3) #Adding Residues ch1.add_residue(res1) ch1.add_residue(res2) ch1.add_residue(res3) #Creating Atoms a1=Atom("OP1", 0.1, 0.2, 0.3, "O") a2=Atom("P", 0.4, -0.5, 0.6, "P") a3=Atom("N1", 0.25, 0.54, 0.23, "N") a4=Atom("C4", 0.21, 0.76, -0.93, "C") #Adding Atoms res1.add_atom(a1) res2.add_atom(a2) res3.add_atom(a4) res3.add_atom(a3) - The following code shows how to access each structural element
print(rna_molecule.get_models()) print(rna_molecule.get_models()[0].get_chains()) print(rna_molecule.get_models()[0].get_chains()[0].get_residues()) print(rna_molecule.get_models()[0].get_chains()[0].get_residues()[0].get_atoms()) - Printing the structure using the
print_all()methodrna_molecule.print_all()provides as output the following format, saved in a file:
1JAT X-RAY DIFFRACTION Homo sapiens Model 1 ATOM 1 OP1 A A 1 0.1 0.2 0.3 O ATOM 2 P U A 2 0.4 -0.5 0.6 P ATOM 3 C4 C A 3 0.21 0.76 -0.93 C ATOM 4 N1 C A 3 0.25 0.54 0.23 N
Small Example integrating RNA moelcules and families
In this example we proceeded with the RNA molecule 7EAF, and retrieved its RNA family which is teh SAM family, by just providing SAM as query and all info is automatically stored within the family object up until its own tree of type PhyloTree (see more in this notebook)
Small example automating creation of all RNA molecules found for a family
In this example, with only information regarding a family, we retrieved all family attributes from database and have a fully functional Family object with name, id, type and tree of type PhyloTree. We also automated the extraction of all RNA molecules found for the family from the PDB database, by first retriving all pdb ids through rfam api, then fetching them through PDB biopython’s api and creating the RNA_Molecule objects which we added as members of the family. Thus a fully declared family with all attributes, more in here